You must be signed in to read the rest of this article.
Registration on CDEWorld is free. You may also login to CDEWorld with your DentalAegis.com account.
As dentists understand more about biofilm biology as it relates to human infections, they are better able to understand how these associations affect healthy and compromised immune systems. The appreciation of bacterial microbiology as it relates to plaque and periodontal diseases elucidates how these bacteria affect decay, periodontal diseases, and host invasion and systemic inflammatory responses. Previously, bacteria were viewed as individual self-sufficient cells that routinely lacked the ability to communicate or organize themselves into groups.1 This view of bacteria—individual cells that did not relate or organize into communities—was called a planktonic growth of bacteria.2 Research of planktonic bacteria was geared to isolate the separate bacteria, grow a pure culture, and identify individual properties to ascertain how to best control these pathogens with antibiotic and antimicrobial medications.3 Recent research has demonstrated that the majority of bacteria live in biofilms, and about 65% of human infections are related to biofilm biophysiology.4 Most infective agents are opportunistic pathogens that form biofilms on tissues or other surfaces, and most biofilms enter the body through either the skin, digestive tract, respiratory system, or mouth.5
Biofilms
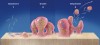
Certain conditions have been discovered that make it conducive for a biofilm to develop from planktonic bacteria. A suitable substrate to which the bacteria can attach with adequate food and nutrients is essential. Bacterial pathogens are able to establish a tight contact with a suitable substrate (tooth surface, eukaryotic cells and tissues) by using cell adhesion receptors: different proteins, immunoglobulin, and other cell and tissue products6 (Figure 1). Planktonic bacteria can initiate biofilm formation through quorum sensing7 and the production of extracellular deoxyribonucleic acid (DNA).8 Different cell-to-cell signal systems are involved in biofilm formation, and Davies et al9 showed that when sufficient food and bacterial population densities existed, these cell-to-cell signals reached a sufficient concentration for activation of genes involved in biofilm differentiation. Yung-Hua et al10 showed that the induction of genetic competence was mediated by quorum sensing and that transformation frequencies were 10 to 600 times higher in biofilms than in planktonic cells.
Biofilms are difficult to deal with through conventional recovery methods (swab, etc), and growth mediums are not sufficient to grow biofilms.11 According to these same authors, a 90% error occurred in bacterial determination when compared with fluorescent in situ hybridization screening. Not only are biofilms hard to culture, but it also is difficult to ascertain the individual bacteria as 50% to 90% of biofilms are comprised of extracellular polymeric substance (EPS).12 Most EPS structure is composed of polysaccharides, which provide both hydrophilic and hydrophobic properties because of the structure of their hydrogen bonding.13 The EPS formation varies spatially and structurally by different organisms and increases with the age of the biofilm.14 The hydrated nature of the EPS contributes to the antimicrobial resistance by impeding the mass transport of antibiotics through the biofilm, most probably by direct bonding properties.15 Colonies containing motile cells can alter their structure through intermixing and migration of cells from one microcolony to another.16 Non-microbial EPS surface components (erythrocytes, fibrin, platelets) also modify the biofilm and may protect the biofilm from polymorphonuclear leukocytes (PMN) and host defenses.17 Exopolysaccharide intercellular adhesions (PIAs) reside in fibrous strands on the bacterial cell surface of some bacteria, and these PIAs inhibit phagocytosis and death by human PMN.18 Organisms within biofilms can share and readily acquire resistance through transfer of resistance plasmids or genetic materials19 (Figure 2).
Immune System Response
The host immune system is able to protect against most planktonic bacteria invasions through one of three mechanisms: (1) phagocytosis of invading microorganisms by blood cells; (2) proteolytic reactions leading to localized responses (clotting, opsonization); or (3) synthesis of antimicrobial peptides.20 Bacteria that live in biofilms are remarkably resistant to host defenses and therapy with conventional antibiotics15,21 because mixed biofilm populations differ from their planktonic counterparts in both genotypic diversity and phenotypic gene expression.22,23 PMN are the first line of defense against infection.24 Biofilms may have an adverse effect on PMN function. Jesatitis et al21 found that neutrophils that settle on biofilms lacked pseudopods, had impaired motility, and became enveloped in the biofilm as planktonic bacteria were released by the biofilm. The PMN also were found to become degranulated with little increase in hydrogen peroxide production and a diminished oxidative potential.21 Evidence by Ward et al25 illustrated that the immune system of a vaccinated rabbit had no effect on the growth of bacteria in biofilms implanted in the animal, which demonstrated that microorganisms are able to detach from biofilms, and could overcome the host immune system and cause an infection. Certain extracellular bacterial components have been found to interfere with host macrophage phagocytic activity.26 Also, biofilm bacteria have been found unable to be eliminated by phagocytosis by opsonized antibodies in cystic fibrosis patients.27 One study found that biofilm cells were less sensitive to death by human PMN cells, leading Yasuda et al28 to conclude that biofilm organisms are resistant to oxygen species produced by PMN, and detached biofilm cells may be able to evade the host phagocytic activity in the bloodstream and initiate a bloodstream infection. Geerts et al29 reported that not only do bacteria enter the bloodstream during mastication, but also endotoxin levels increased in the bloodstream fourfold after mastication. They further found that endotoxin levels of severe periodontitis patients were greater than mild or moderate disease states.29 Kinane et al30 reported on an increased incidence of bacteremia induced from conventional periodontal procedures. Forner et al31 stated that the crucial nature of periodontal treatment is the prevention of bacteremia associated with oral procedures, whereas Misra et al32 stipulated that there is an increased possibility of bacteremia being more frequent and affecting children with congenital heart defects and subsequent endocarditis.
The host periodontal sulcus responds in predictive ways to associated plaque in localized periodontitis. Verderame et al33 discovered epithelial cavitations and ulcerations in response to the microorganisms entrapped in the region. The development of epithelial breakdown and ulcerations might help explain findings by Matheny et al,34 who, while evaluating the microcirculatory dynamics associated with human gingivitis, discovered a significant increase in the number of blood vessels visible in microscopic fields. Kerdvongbundit et al35 evaluated inflammatory changes in the microcirculatory and micro-morphologic dynamics of human gingiva before and after conventional treatment (scaling and root planing). Blood flow measured with laser doppler flowmetry demonstrated a statistically significant blood flow increase when the gingival tissues were inflamed. These returned to normal after treatment and remained stable for 3 months posttreatment. Giorgio Cimasoni of the University of Geneva School of Dentistry Department of Periodontics demonstrated the close proximity and relationship of the bloodstream to the periodontal pocket (Figure 3). The increased exposure of the bloodstream during infection helps explain how perio-pathogenic cells might become involved systemically or have systemic effects on the host immune system as the host responds to these pathogens.
Research is showing that certain of these pathogens are able to invade human cells,36 thus making pathogen recognition and control more difficult.37 This cellular invasion and difficulty in recognition places patients with a medical or immune compromise at greater risk from biofilm pathogens.38 Lu and Jacobson39 demonstrated that patients with immunodeficiencies, therefore, are more susceptible to infection and experience a greater degree of infection than patients with competent immune systems. Primary immunodeficiencies include humor immunities (affecting B-cell differentiation or antibody production), T-cell defects, combined B- and T-cell defects, phagocytic disorders, and complement deficiencies. These multiple disorders of the host immune system often involve multiple infections with unusual or opportunistic organisms. These infections occur despite aggressive treatments, with the host experiencing a failure to thrive or grow, which often is associated with a positive family history.40
How Normal Host Responses Challenge Biofilm Infections
Patients with a normal immune system are able to counter most planktonic bacterial infections on their mucosal surfaces.41 The bone marrow is the ultimate source of blood cells, including those destined to become immune cells. The lymphocytes comprise a majority of the immune cells that originate from stem cells and are comprised of T cells, which mature in the thymus, and B cells.42 Lymphocytes travel via the bloodstream and also through the lymphatic vessels as fluid and cells are exchanged between these two systems.43 Within the lymphatic system are small lymph nodes that contain specialized compartments where immune cells encounter foreign particles.44 To work effectively, the cells of the immune system must communicate either by physical contact or by chemical messengers, such as cytokines.45
B cells and T cells are the main type of lymphocytes. B cells work chiefly by fabricating and secreting antibodies in response to specific antigens, and these antibodies attach to the antigens and mark the antigen for destruction.46 Antibodies belong to a large group of molecules known as immunoglobulins, which play different roles in immune system function. Immunoglobulin G (IgG) works to coat microbes, accelerating their recognition and uptake by other cells in the immune system.47 Immunoglobulin M (IgM) is effective in killing bacteria, whereas immunoglobulin A (IgA) functions via secretions in the digestive tract, tears, and saliva to help guard against entry infections.48 These and a host of other immunoglobulins are generally effective in helping manage the host challenge to bacterial infections. According to the National Cancer Institute, in patients with normal immune system antibodies, the antibodies bind to and inactivate bacterial toxins. The antibodies bind to the antigen and make it recognizable to phagocytic cells (opsonization), activating the complement cascade, blocking the antigen from cell invasion, and binding to the cell, which makes it possible for killer immune cells to destroy the pathogens.49 Results of activation of the complement cascade include stimulating mast cells and basophils to release granulocytic chemicals, neutrophil attractants, and opsonizing compounds, and to generate membrane attractant complexes (C1q, C3, C4, C5, C5-9 [membrane attack complex (MAC)]), factors B and Fb, factor H, and properdine, some of which serve to break down pathogen membranes.50
Unlike B cells, T cells do not recognize antigens, but their surfaces contain antibody-like receptors that are involved in immune response. Memory T cells are required to maintain immunity, while regulatory T cells help keep the immune system in check to prevent inflammation and autoimmunity.51 Killer T cells directly attack foreign cells, using molecules on their surface to recognize small fragments (antigens) and launch an attack to kill the foreign cell.52 These surface receptors (major histocompatibility complex [MHC]) molecules are proteins used for non-self-recognition. These MHC proteins present significant problems with transplants as almost all cells are covered with MHC proteins and the donor/recipient pattern must be a close match.53 Natural killer (NK) cells are armed with granules filled with potent chemicals that are attracted to cells lacking self-MHC molecules and attach to other types of foreign cells. NK cells have the potential to bind to many types of foreign cells, and then deliver their chemical barrage to kill the pathogens.54
T cell cytokines help regulate monocyte/macrophage function.55 Monocytes are phagocytic cells that circulate in the blood and appear to respond to specific cytokines (interleukin [IL]-10), which cause the cells to migrate into tissues where they develop into macrophages.56 Macrophages, in response to a host of signals, scavenge and rid the body of worn out cells (PMN) and other debris.57 The National Cancer Institute has stated that granulocytes that include basophils produce chemicals, such as histamines, and are able to destroy planktonic bacteria. However, granulocytes also contribute to inflammation and some allergic reactions.
These same authors stated that eosinophils release granulocytic chemicals into surrounding tissues to destroy pathogens,49 while neutrophils are phagocytic cells that are often the first line of defense as these cells respond to cytokines as well as produce cytokines and ingest planktonic bacteria, and use a series of enzymes and hydrogen peroxide (superoxide) to kill the ingested pathogens.58 One of the products produced in a PMN is lactoferrin, which is a multifunctional, antimicrobial (bactericidal) protein. Lactoferrin also is produced on acinar cells of the pancreas, stomach, salivary glands, and other organs.59
For an immune system to function in a normal manner, its components must be able to create and exchange proteinaceous cytokines, which enable the system to communicate. Some cytokines activate certain immune cells, whereas others turn off specific cells. When stimulated by infection, T helper cells fabricate IL-2, which serves to increase the number of infection-fighting cells and causes them to mature.60 Specific cytokines attract specific cells, whereas injured cells also produce chemokines which attract or stimulate immune cells and/or are factors in inflammation and the regulation of immune responses.61 Complement proteins are free-circulating inactive agents that serve to complement antibody-coating antigen complexes. When activated (typically by an antibody), a chain reaction of complements occurs with the end result usually puncturing a hole in the pathogen cell wall, thus increasing the pathogen susceptibility to phagocytosis and/or activation of attractants for phagocytosis.62
Biofilms complicate the immune system recognition and control. Biofilms are highly structured communities of bacteria usually embedded in an exopolysaccharide and proteinaceous matrix that protects the community from antibiotics and the immune system18 (Figure 4). Leid et al63 demonstrated that the exopolysaccharide matrix of Pseudomonas aeruginoa caused a biofilm to be refractory to the host immune system, but cells unable to form the biofilm matrix were susceptible to the host immune system. Wagner et al64 found that when PMN were exposed to biofilms, they experienced a significant alteration of function as they underwent transdifferentiation. These authors found that PMN lost the ability to respond to CD62L and the up-regulation of CD14 as well as the expression of CD83, which resulted from the effects of the infection on the host cells. This finding helped explain how biofilm-affected PMN lost their chemotactic activity, while the production of superoxide and opsonized chemicals (cytotoxic, proteolytic, and collagenolytic) potentially continued and was enhanced. This combination provides a possible explanation of how the affected PMN might contribute to tissue destruction and eventually to tissue lyses.
Conclusion
Biofilm disease affects the host immune system in a variety of ways. Various biofilm components cause different immune system responses. Treatment of the biofilm is an integral aspect to decrease the immune system responses, along with decreasing the host systemic inflammation.
References
1. Greenberg EP. Bacterial communication; tiny teamwork. Nature. 2003;424(6945):134.
2. Marxsen J. Studies on the structure and function of planktonic bacteria in two small West German streams [abstract]. Verh Internat Verein Limn. 1978;20:1516.
3. Teacher’s Guide: Antibiotics in Action. Biology Activity: Culturing Bacteria, Isolation and Identification. Philadelphia, PA: The Chemical Heritage Foundation; 2002.
4. Costerton JW, Stewart PS, Greenberg EP. Bacterial biofilms: a common cause of persistent infections. Science. 1999;284(5418);1318-1322.
5. Jakob HG, Morneff-Lipp M, Bach A, et al. The endogenous pathway is a major route for deep sternal wound infection. Eur J Cardiothorac Surg. 2000;17(2):154-160.
6. Hauck CR. Cell adhesion receptors—signaling capacity and exploitation by bacterial pathogens. Med Microbiol Immunol. 2002;191(2):55-62.
7. Sritharan M, Sritharan V. Emerging problems in the management of infectious diseases: the biofilms. Indian J Med Microbiol. 2004;22(3):140-142.
8. Whitchurch C B, Tolker-Nielsen T, Ragas PC, et al. Extracellular DNA required for bacterial biofilm formation. Science. 2002;295(5559):1487-1493.
9. Davies DG, Parsek MR, Pearson JP, et al. The involvement of cell-to-cell signals in the development of a bacterial biofilm. Science. 1998;280(5361):295-298.
10. Yung-Hua L, Lau PCY, Lee JH, et al. Natural genetic transformation of Streptococcus mutans growing in biofilms. J Bacteriol. 2001;183(3):897-908.
11. Veeh RH, Shirtliff ME, Petik JR, et al. Detection of Staphylococcus aureus biofilm on tampons and menses components. J Infect Dis. 2003;188(4):519-30.
12. Flemming H-C, Wingender J, Griegbe, et al. Physico-chemical properties of biofilms. In: Evans LV, ed. Biofilms: Recent Advances in Their Study and Control. Amsterdam: Harwood Academic Publishers; 2000:19-34.
13. Sutherland I. Biofilm exopolysaccharides; a strong and sticky framework. Microbiology. 2001;147(Pt 1):3-9.
14. Leriche V, Sibille P, Carpentier B. Use of an enzyme-linked lectin-sorbent assay to monitor the shift in polysaccharide composition in bacterial biofilms. Appl Environ Microbiol. 2000;66(5):1851-1856.
15. Donlan RM. Role of biofilms in antimicrobial resistance [published erratum appears in: ASAIO J. 2001;47(1):99]. ASAIO J. 2000;46(6):S47-S52.
16. Durack DT. Experimental bacterial endocarditis. IV. Structure and evolution of very early lesions. J Pathol. 1975;115(2):81-89.
17. Tunney MM, Jones DS, Gorman SP. Biofilm and biofilm-related encrustations of urinary tract devices. In: Abelson JN, Simon MI, Doyle RJ, eds. Methods in Enzymology, Vol. 310: Biofilms. San Diego, CA: Academic Press; 1999:558-566.
18. Vuong C, Voyich JM, Fischer ER, et al. Polysaccharide intercellular adhesin (PIA) protects Staphylococcus epidermidis against major components of the human innate immune system. Cell Microbiol. 2004;6(3):269-275.
19. Martinez LR, Casadevall A. Cryptococcus neoformans cells in biofilms are less susceptible than planktonic cells to antimicrobial molecules produced by the innate immune system. Infect Immun. 2006;74(11):6118-6123.
20. Hoffmann JA, Kafatos FC, Janeway CA Jr, et al. Phylogenetic perspectives in innate immunity. Science. 1999;284(5418):1313-1318.
21. Jesatitis AJ, Franklin MJ, Berglund D, et al. Compromised host defense of Pseudomonas aeruginoa biofilms: characterization of neutrophils and biofilm interactions. J Immunol. 2003;171(8):4329-4339.
22. Boles BR, Thoendel M, Singh PK. Self-generated diversity produces “insurance effects” in biofilm communities. Proc Natl Acad Sci U S A. 2004;101(47):16630-16635.
23. Sauer K, Camper AK, Ehrlich GD, et al. Pseudomonas aeruginosa displays multiple phenotypes during development as a biofilm. J Bacteriol. 2002;184(4):1140-1154.
24. Components of the immune system. November 2005. The Merck Manuals Online Medical Library. Available at: http://www.merck.com/mmpe/sec13/ch163/ch163b.html. Accessed Sep 29, 2008.
25. Ward KH, Olson ME, Lam K, et al. Mechanism of persistent infection associated with peritoneal implants. J Med Microbiol. 1992;36(6):406-403.
26. Shiau AL, Wu CL. The inhibitory effect of Staphylococcus epidermidis slime on the phagocytosis of murine peritoneal macrophages is interferon-independent. Microbiol Immunol. 1998;42(1):33-40.
27. Meluleni GJ, Grout M, Evans DJ, et al. Mucoid Pseudomonas aeruginosa growing in a biofilm in vitro are killed by opsonic antibodies to the mucoid exopolysaccharide capsule but not by antibodies produced during chronic lung infection in cystic fibrosis patients. J Immunol. 1995;155(4):2029-3208.
28. Yasuda H, Ajiki Y, Aoyama J, et al. Interaction between human polymorphonuclear leucocytes and bacteria released from in vitro bacterial biofilm models. J Med Microbiol. 1994;41 (5):359-367.
29. Geerts SO, Nys M, De MP, et al. Systemic release of endotoxins induced by gentle mastication: association with periodontitis severity. J Periodontol. 2002;73(1):73-78.
30. Kinane DF, Riggio MP, Walker KF, et al. Bacteraemia following periodontal procedures. J Clin Periodontol. 2005;32(7):708-713.
31. Forner L, Larsen T, Kilian M, et al. Incidence of bacteremia after chewing, tooth brushing and scaling in individuals with periodontal inflammation. J Clin Periodontol. 2006;33(6):401-407.
32. Misra S, Percival RS, Devine DA, et al. A pilot study to assess bacteraemia associated with tooth brushing using conventional, electric or ultrasonic toothbrushes. Eur Arch Paediatr Dent. 2007;8(Suppl 1):42-45.
33. Verderame RA, Cobb CM, Killoy WJ, et al. Scanning electron microscopic examination of pocket wall epithelium and associated plaque in localized juvenile periodontitis. J Clin Periodontol. 1989;16(4):234-241.
34. Matheny JL, Abrams H, Johnson DT, et al. Microcirculatory dynamics in experimental human gingivitis. J Clin Periodontol. 1993;20(8):578-583.
35. Kerdvongbundit V, Vongsavan N, Soo-Ampon S, et al. Microcirculation and micromorphology of healthy and inflamed gingivae. Odontology. 2003;91(1):19-25.
36. Rudney JD, Chen R, Sedgewick GJ. Intracellular Actinobacillus actinomycetemcomitans and Porphyromonas gingivalis in buccal epithelial cells collected from human subjects. Infect Immun. 2001;69(4);2700-2707.
37. Zhang Y, Wang T, Chen W, et al. Differential protein expression by Porphyromonas gingivalis in response to secreted epithelial cell components. Proteomics. 2005;5(1):198-211.
38. Sullivan, KJ, Goodwin SR, Sandler E, et al. Critical care of the pediatric hematopoietic stem cell transplant recipient in 2005. Pediat Transplant. 2005;9(Suppl 7):12-24.
39. Lü FX, Jacobson RS. Oral mucosal immunity and HIV/SIV infection. J Dent Res. 2007;86(3):216-226.
40. Cooper MA, Pommering TL, Korányi K. Primary immunodeficiencies. Am Fam Physician. 2003;68(10):2001-2008.
41. Ganz T. Antimicrobial proteins and peptides in host defense. Semin Respir Infect. 2001;16(4):4-10.
42. Geissmann F, Jung S, Littman DR. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity. 2003;19(1):71-76.
43. Michel CC. Starling: the formulation of his hypothesis of microvascular fluid exchange and its significance after 100 years. Exp Physiol. 1997;82(1):1-30.
44. Mc Dowell J, Windelspecht M. The Lymphatic System. Westport, CT: Greenwood Press; 2004.
45. Cuk M, Radoseviç-Stasiç B, Milin C, et al. Lymphoid system as a regulator of morphostasis and hormonal modulation of these functions. Ann N Y Acad Sci. 1987;496(1):104-107.
46. Shachar I, Flavell RA. Requirement for invariant chain in B cell maturation and function. Science. 1996;274(5284):106-108.
47. IgG. March 2008. Dalhousie University Faculty of Medicine Immunology Bookcase. Available at: http://pim.medicine.dal.ca/igg.htm. Accessed Sep 30, 2008.
48. Norrby-Teglund A, Ihendyane N, Kansal R, et al. Relative neutralizing activity in polyspecific IgM, IgA, and IgG preparations against Group A streptococcal superantigens. Clin Infect Dis. 2000;31(5):1175-1182.
49. Oppenheim JJ, Biragyn A, Kwak LW, et al. Role of antimicrobial peptides such as defensins in innate and adaptive immunity. Ann Rheumatol Dis. 2003;62(Suppl 2):ii17-ii21.
50. Nelson KC, Zhao M, Schroeder PR, et al. Role of different pathways of the complement cascade in experimental bullous pemphigoid. J Clin Invest. 2006;116(11):2892-2890.
51. Akbar AN, Vukmanovic-Stejic M, Taams LS, et al. The dynamic co-evolution of memory and regulatory CD4+ T cells in the periphery. Nat Rev Immunol. 2007;7(3):231-237.
52. Lebbink RJ, Meyaard L. Non-MHC ligands for inhibitory immune receptors: Novel insights and implications for immune regulation. Mol Immunol. 2007;44(9):2153-2164.
53. Figueiredo C, Horn PA, Blasczyk R, et al. Regulating MHC expression for cellular therapeutics. Transfusion. 2007;47(1):18-27.
54. Castriconi R, Dondero A, Cantoni C, et al. Functional characterization of natural killer cells in type I leukocyte adhesion deficiency. Blood. 2007;109(11):4873-4881.
55. Stout RD, Suttles J. T cell signaling of macrophage function in inflammatory disease. Front Biosci. 1997;2:197-206.
56. Prasse A, Germann M, Pechkovsky DV, et al. IL-10 producing monocytes differentiate to alternatively activated macrophages and are increased in atopic patients. J Allergy Clin Immunol. 2007;119(2):464-471.
57. Myung PS, Clements JL, White DW, et al. In vitro and in vivo macrophage function can occur independently of SLP-76. Int Immunol. 2000;12(6):887-897.
58. MacIvor DM, Shapiro SD, Pham CT, et al. Normal neutrophil function in cathepsin G-deficient mice. Blood. 1999;94(12):4282-4293.
59. Orsi N. The antimicrobial activity of lactoferrin: current status and perspectives. Biometals. 2004;17(3):189-196.
60. Chen CY, Gherzi R, Andersen JS, et al. Nucleolin and YB-1 are required for JNK-mediated interleukin-2 mRNA stabilization during T-cell activation. Genes Dev. 2000;14(10): 1236-1248.
61. Allen SJ, Crown SE, Handel TM. Chemokine: receptor structure, interactions and antagonism. Annu Rev Immunol. 2007;25:787-820.
62. Dumestre-Pérard C, Doerr E, Colomb MG, et al. Involvement of complement pathways in patients with bacterial septicemia. Mol Immunol. 2007;44(7):1631-1638.
63. Leid JG, Wilson CJ, Shirtliff ME, et al. The exopolysaccharide alginate protects Pseudomonas aeruginosa biofilm bacteria from IFN-gamma-mediated macrophage killing. J Immunol. 2005;175(11):7512-7518.
64. Wagner C, Kaksa A, Müller W, et al. Polymorphonuclear neutrophils in posttraumatic osteomyletitis: cells recovered from the inflamed site lack chemotactic activity but generate superoxides. Shock. 2004;22(2):108-115.
About the Authors
Duane C. Keller, DMD
President
Keller Professional Group
President/CEO
Perio Protect, LLC
St. Louis, Missouri
J. William Costerton, PhD
Biofilm Research
Center for Genomic Sciences
Allegheny-Singer Research Institute
Pittsburgh, Pennsylvania